Auxiliary Programs for multiX
gv
a viewer program for postscript graphic files. Note: ps files can be read and edited. ps files are ready for possible inclusion into publications and can be printed on any (postscript) printer. gv is used in the present context, to view spectral output from multiX as processed by plt_mult. AMOE_mult also produces ps graphics output.
installation: sudo apt install gv
use: gv graph.ps
xbs
a versatile and sleek viewer for 3-d ball-stick structures. Uses l0.bs input files prepared by mk_bs.
installation1: sudo apt-get update
installation2: sudo apt-get install xbs
use: xbs l0
screenshot
a standard linux system program to grab graphical info on the screen and produce a pnm file.
plt_mult
a program to prepare a graph.ps file as postscript graphics showing a spectrum from a multiX spect file.
use:
plt_mult xas
plt_mult xmld or plt_mult xld (peak normed to 1)
plt_mult xmcd or plt_mult spect-xmcd.dat (to show Carra integral functions in addition)
plt_mult ins
plt_mult any_filename
plt_mult offers interactive input for plot refinement options, including Gaussian convolution.
It can also prepare spectral graphics files from any multicolumn spectral file (it tries to skip header) with energy in the first column. use:
When working on a file with uneven energy spacing, it prepares an equi_en file with
interpolated function values with constant energy steps according
to the minimum energy step of the original file.
Equi-energy step tabulations are required by AMOE_mult as a target function for optimization.
The equi_en file will typically be trimmed to a tighter energy range, and renamed for use as reference spectral data in an optimization and refinement of semiempirical paramters used in the multiX model.
There are options for background subtraction when preparing the equi_en file:
"1" subtracts a linear background setting the end points of the tabulation to zero.
"2" subtracts a quadratic background by averaging 9 points of the tabulation left, middle and right as reference values to be zeroed. The intent is that the middle point falls into 'background' between L2/L3 or M4/M5.
"L0...L40" allows to use a polynomial fit for background subtraction.
cif_construct
constructs a DMol3 type crystal coordinates file in Bohr length units from a cif file
use: cif_construct file.cif |tee i_from_cif
note: some cif files contain fractionally occupied sites. All sites are shown in the cif_construct output.
The crystal lattice info in i_from_cif can be visualized using the mk_bs and xbs programs.
cif_construct simply constructs all the lattice positions for all given sites irrespective of possibly fractional occupation.
mk_lig
extracts a cluster of ligand sites around the first atomic site in the crystal file
usually it is necessary to edit the i_from_cif crystal file to have the desired atomic site listed first in i_modified
use: mk_lig i_modified 8 |tee i8_ligands
where the number is the cut off radius in Angs for the cluster
output: ligands.dat file, which could be used directly for a multiX calculation by the xtal_file command line: xtal_file ligands.dat.
However, we prefer now to use an editor and prepare an input section bracketed between begin_xtal and end_xtal as shown in most examplesa.
The output also contains a DMol3 style cluster input including the central atom. It also contains a sketchy section as a start to produce a FEFF8 input file.
The ligands.dat file, or the similar output file i8_ligands can be visualized using the mk_bs and xbs programs.
mk_bs
prepares a l0.bs file from DMol3 style coordinate data.
The l0.bs file serves for quick structural viewing with the handy XBS ball stick visualizer program.
use: mk_bs ligands.dat
or: mk_bs i_from_cif
AMOE_mult
finds semi-empirical multiX parameters for a better fit to an experimental spectrum.
Patiently minimizes the RMS of the residual difference theo-exp under user control using the simplex method
for one or several model spectra to given experimental spectra.
use: nohup AMOE_mult &
Any numerical parameter in a multiX INPUT file can be subject to bounded variation by tagging it as a variable in a prototype INPUT file for multiX an defining its bounds in the control file input_amoe.
full downloadable set of commented example files:
-
input_amoe ,
i_XAS0 ,
XAS_SrTiO3 ,
xas_SrTiO3_fit.png ,
nohup.out ,
comments.
example files for fitting multiple spectra with multiple models for each spectrum, due to chemically inequivalent sites"
-
input_amoe ,
i_XMCD_g ,
i_XMCD_g4c ,
i_XMLD,
i_XMLD_4c,
fit_of_4_spectra,
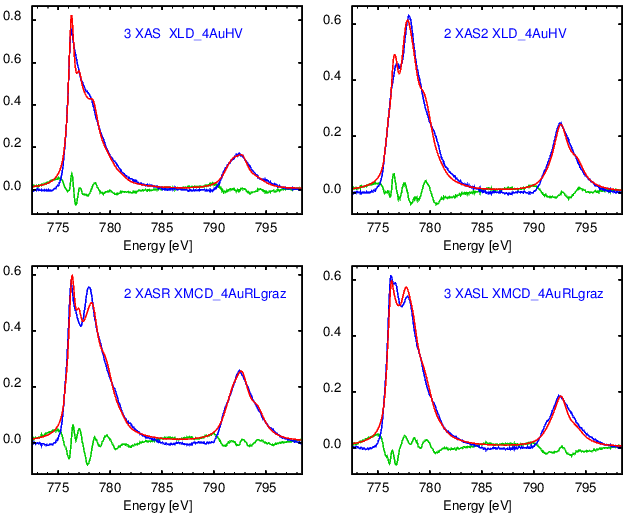
Note that the measured spectra and not the difference spectra XMCD XLD XMLD are chosen to be fitted.
The current AMOE_mult keeps multix spectral files for each spectral contribution as s_.... files ,
it also keeps the corresponding multiX output files as u_... files .
The latest found optimal fit parameters are kept in the inp_lo file .
The latest established optimal fit is documented in the graph.ps file and eventually overwritten with experimental spectrum: blue, fit: red and residual: green.
back to index
if your project could profit from another auxiliary program, contact BD, it may already have been developed.
rev 250417BD
{kind=link}
{kind=link}